
Neuropatia delle Piccole Fibre Bambini e Adolescenti Dolore e Disautonomia | Prove Scientifiche di SFPN sin dal 2013 per Sindromi Inspiegabili di Dolore Diffuso ad Insorgenza Giovanile: neuropatia-delle-piccole-fibre-bambini-e-adolescenti-dolore-e-disautonomia
Neuropatia delle Piccole Fibre Bambini e Adolescenti Dolore e Disautonomia: La Traduzione di un articolo pubblicato su Pediatrics 2013, Autori Oaklander e Kein del Massachusetts General Hospital, luminari per la Neuropatia delle Piccole Fibre (Small Fiber Neuropathy, in questo studio definita più ampiamente Small Fiber Polyneuropathy, Polineuropatia delle Piccole Fibre). Questo studio ha analizzato i risultati di più test oggettivi per identificare una causa potenzialmente comune per sindromi di Dolore Cronico Diffuso (CWP) ad esordio infantile: la SFPN. Estende il range di età della SFPN alla prima infanzia e caratterizza la presentazione pediatrica. Prima della traduzione integrale, un riassunto in 10 punti, estratto dall’articolo originale : neuropatia-delle-piccole-fibre-bambini-e-adolescenti-dolore-e-disautonomia
- Questa serie di casi fornisce nuove ipotesi sulle sindromi di inspiegabile Dolore Cronico Diffuso ad insorgenza nell’infanzia, implicanti SFPN acquisita, una diagnosi biologicamente plausibile non precedentemente riconosciuta nei bambini.
- Il riconoscimento della SFPN a esordio giovanile dovrebbe aiutare pazienti, famiglie e medici suggerendo un percorso razionale per la valutazione e il trattamento diagnostico. Avere una diagnosi specifica da testare e curare, quando presente, può ridurre test e trattamenti inefficaci, costosi e potenzialmente dannosi e consentire test oggettivi e il trattamento definitivo di alcuni pazienti.
- La somministrazione cronica di farmaci per il dolore palliativo ha effetti avversi medici e sociali che sono ancora meno accettabili nei bambini che negli adulti.
- Queste sindromi giovanili di Dolore Cronico Diffuso sempre più frequenti, spesso distruggono intere famiglie e interferiscono con l’educazione e lo sviluppo dei bambini
- L’età media all’esordio dei sintomi era di 12,3 ± 5,7 anni; l’età alla presentazione era in media di 20,8 ± 9,1 anni. I dati suggeriscono che la SFPN può svilupparsi anche nei bambini in età prescolare e che la SFPN ad esordio giovanile può persistere per decenni nell’età adulta.
- La SFPN di solito inizia negli assoni più lunghi, causando dolore distale (alle estremità, mani e piedi) o in prevalenza distale (nei piedi). Alcuni pazienti sviluppano sintomi a chiazze (persino sulle guance e orecchie) o prevalentemente prossimali, attribuiti all’attacco prossimale sui corpi cellulari neuronali (ganglionopatia/neuronopatia). Poiché le piccole fibre innervano densamente e regolano anche il tono dei microvasi, un aspetto anomalo (ad es. edemi, cambiamenti di colore) è comune nelle aree interessate. Sono stati identificati tutti i caratteristici pattern temporali (acuto/fulminante versus cronico) e spaziali (distale versus a chiazze/prossimale) delle polineuropatie.
- Gli esami generali hanno identificato costantemente tachicardia sinusale e pressione arteriosa anomala, di solito ipotensione ortostatica. Diversi pazienti hanno documentato vesciche neurogene episodiche. Queste erano spesso sugli arti, anche nel cavo orale e sul viso. I pazienti occasionali avevano riflessi pupillari pigri.
- Le sindromi di Dolore Cronico Diffuso spesso includono altri disturbi inspiegabili tra cui vertigini, stanchezza, mal di testa e nausea. Più della metà dei pazienti giovani con capogiri attribuiti alla Sindrome da Tachicardia Ortostatica Posturale (Postural Orthostatic Tachycardia Syndrome, POTS) aveva anche cefalea cronica, dolore addominale (inclusa grave stipsi) e/o stanchezza suggerendo una possibile eziologia comune. Con sintomi disautonomici onnipresenti, segni e anomalie dei test, la SFPN giovanile integra componenti del dolore sia autonomo che neuropatico. Inaspettatamente, il 63% dei pazienti soffriva di mal di testa cronico.
- La maggior parte dei pazienti era malata moderatamente o gravemente, dimostrato dalle loro vaste indagini/valutazioni mediche presso i principali centri medici accademici. Il 68% era stato ricoverato in ospedale e il 68% aveva chiesto di lasciare la scuola o il lavoro. Non sono state identificate altre diagnosi patogenetiche che spiegassero il loro dolore diffuso (ad es., Artrite, miopatia). L’etichetta sindromica più comune all’ingresso nello studio era la fibromialgia; altri includevano disordine funzionale, sensibilizzazione centrale, sindrome da amplificazione del dolore, stanchezza cronica, sindrome del dolore miofasciale e malattia di Lyme cronica sieronegativa. Le etichette sindromiche specifiche per organo comprendevano POTS, intestino irritabile, dispepsia funzionale, emicrania addominale e cefalea cronica quotidiana. Quattro pazienti avevano anche sindrome dell’ovaio policistico e 3 avevano la sindrome di Ehlers-Danlos. Le diagnosi psichiatriche (somatizzazione, conversione) erano state spesso considerate, sebbene fosse stata documentata solo 1 malattia psichiatrica, in particolare la depressione maggiore attribuita al dolore incessante.
- La diagnosi di SFPN è difficile perché i segni familiari di neuropatia delle grandi fibre sono assenti o minimi e il test elettrodiagnostico è insensibile. I test raccomandati per la diagnosi obiettiva di SFPN sono stati applicati qui: biopsia cutanea specificamente distale della gamba, immunomarcata per rivelare le fibre nervose epidermiche nocicettive (Epidermal Nerve-Fibers, ENF) (raccomandazione di livello C dell’American Academy of Neurology, raccomandazione di livello A della Federazione Europea delle Società di Neurologia), e test di funzionalità autonomica (Autonomic Function Testing, AFT). Questo consiste in 4 test validati della funzione cardiovagale, adrenergica e sudomotoria delle piccole fibre (raccomandazione di livello B dell’American Academy of Neurology). La biopsia cutanea neurodiagnostica, i test di funzionalità autonomica e la biopsia del nervo consentono una diagnosi obiettiva.
La seguente traduzione è puramente a scopo divulgativo e non è in alcun modo a scopo di lucro. Qualsiasi utilizzo a scopo di lucro non è autorizzato. Il testo di seguito non sostituisce in alcun modo fonti mediche ufficiali, né il lavoro del medico. Rivolgersi sempre al proprio medico e fare sempre riferimento a testi e fonti originali.
Prove di Polineuropatia delle Piccole Fibre in Inspiegabili Sindromi di Dolore Diffuso ad Insorgenza Giovanile. Neuropatia delle Piccole Fibre Bambini e Adolescenti
Titolo Originale: Evidence of small-fiber polyneuropathy in unexplained, juvenile-onset, widespread pain syndromes. Neuropatia delle Piccole Fibre Bambini e Adolescenti
Autori: Oaklander AL, Klein MM.
Pubblicato su: Pediatrics. 2013 Apr;131(4):e1091-100. doi: 10.1542/peds.2012-2597
Abstract: https://www.ncbi.nlm.nih.gov/pubmed/23478869
Abstract
Obiettivi: Abbiamo testato l’ipotesi che la Polineuropatia delle Piccole Fibre (Small-Fiber Polyneuropathy, SFPN) acquisita, non precedentemente descritta e definita nelle sue caratteristiche nei bambini, contribuisca ad inspiegabili sindromi pediatriche di dolore diffuso.
Metodi: Quarantuno pazienti consecutivi valutati per dolore diffuso e inspiegabile, con esordio prima dei 21 anni di età, sono stati analizzati approfonditamente mediante le cartelle cliniche relative a test diagnostici oggettivi per SFPN (biopsia cutanea neurodiagnostica, biopsia del nervo e test di funzionalità autonomica), oltre alle storie cliniche, sintomi, segni, altri test e trattamenti. Volontari sani e demograficamente associati hanno fornito controlli normali per i test della SFPN.
Risultati: L’età alla comparsa della malattia è stata in media di 12,3 ± 5,7 anni; il 73% di questo campione polietnico era di genere femminile (P = 0,001). Il 68% era disabile cronico e il 68% era stato ricoverato in ospedale. Il test obiettivo ha diagnosticato “SFPN definita” nel 59%, “probabile” SFPN nel 17% e “possibile SFPN” nel 22%. Solo 1 su 41 aveva risultati di test per SFPN completamente normali. Il 98% dei pazienti presentava altri disturbi somatici compatibili con la disautonomia data dalla SFPN (90% cardiovascolare, 82% gastrointestinale e 34% urologica), l’83% ha riportato stanchezza cronica e il 63% presentava cefalea cronica.
Gli esami neurologici hanno identificato sensazioni ridotte nel 68% e anomalie vasomotorie nel 55%, incluso il 23% con eritromelalgia. Ricerche esaustive per la causa della SFPN hanno identificato solamente una storia di malattie autoimmuni nel 33% dei casi e marcatori sierologici di disordini immunologici nell’89%. Il trattamento con corticosteroidi e/o immunoglobuline per via endovenosa ha portato beneficio obiettivo e soggettivo nell’80% dei pazienti (12/15).
Conclusioni: Più della metà di una vasta serie di pazienti con dolore cronico diffuso inspiegabile nell’infanzia, ha incontrato criteri diagnostici rigorosi, multitest, per SFPN, che estende la fascia di età della SFPN acquisita alla prima infanzia. Alcuni casi sono risultati immuno-mediati e sono migliorati con terapie immunomodulanti.
Cosa è noto su questo argomento:
Le sindromi dolorose diffuse acquisite dei giovani sono comuni, invalidanti, di solito inspiegabili e non trattabili. La Polineuropatia delle Piccole Fibre causa dolore diffuso e disturbi multisistemici negli anziani. Alcune cause sono trattabili. La biopsia cutanea neurodiagnostica, i test di funzionalità autonomica e la biopsia del nervo consentono una diagnosi obiettiva.
Cosa aggiunge questo studio:
Identifica la Polineuropatia delle Piccole Fibre “definita” (nel 59%) e “probabile” (nel 17%) tra 41 pazienti giovani con dolore diffuso, inspiegabile, insorto nell’infanzia. Definisce le caratteristiche cliniche, la diagnostica e le opzioni di trattamento di questa nuova malattia. Alcuni casi sono risultati immunomediati e hanno risposto alle terapie immunomodulanti.
Articolo Originale Integrale: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4074641/
Articolo Integrale Tradotto: Neuropatia delle Piccole Fibre Bambini e Adolescenti
Titolo Originale: Evidence of small-fiber polyneuropathy in unexplained, juvenile-onset, widespread pain syndromes.
Introduzione:
Sindromi che includono il Dolore Diffuso Cronico inspiegabile (Chronic Widespread Pain, CWP) sono diffuse e problematiche nei bambini e negli adulti. L’American College of Rheumatology definisce il CWP come un dolore che colpisce l’assiale (la parte assiale del corpo, busto e testa N.d.T.), più la parte superiore e inferiore del corpo, più la sinistra e la destra, che durano ≥ 3 mesi.1 L’incertezza sull’eziologia e sulla patogenesi del CWP preclude un trattamento efficace con terapie modificanti la malattia, e la somministrazione cronica di farmaci per il dolore palliativo ha effetti avversi medici e sociali che sono ancora meno accettabili nei bambini che negli adulti.
Queste sindromi di CWP giovanili sempre più riconosciute spesso distruggono intere famiglie e interferiscono con l’educazione e lo sviluppo dei bambini.2-4 Le sindromi di CWP spesso includono altri disturbi inspiegabili tra cui vertigini, stanchezza, mal di testa e nausea. Più della metà dei pazienti giovani con capogiri attribuiti alla Sindrome da Tachicardia Ortostatica Posturale (Postural Orthostasis Tachycardia Syndrome, POTS) aveva anche cefalea cronica, dolore addominale e/o stanchezza,5 suggerendo una possibile eziologia comune.
La Polineuropatia a prevalenza delle Piccole Fibre (Small-Fiber Polyneuropathy, SFPN) è un substrato biologico plausibile per il CWP. La SFPN riguarda danni diffusi che interessano prevalentemente gli assoni periferici di piccolo diametro non mielinizzati (fibre C) o sottilmente mielinizzati (δ-A) che proteggono l’organismo segnalando il dolore al contatto dannoso. La SFPN di solito inizia negli assoni più lunghi, causando dolore distale o in prevalenza distale (nei piedi). Alcuni pazienti sviluppano sintomi a chiazze o prevalentemente prossimali, attribuiti all’attacco prossimale sui corpi cellulari neuronali (ganglionopatia/neuronopatia; Fig 2A).6
Poiché le piccole fibre innervano densamente e regolano anche il tono dei microvasi (microcircolazione N.d.T), è comune un aspetto anomalo nelle aree interessate (ad es. edemi, cambiamenti di colore).7 Le piccole fibre erano precedentemente dicotomizzate come somatiche rispetto a quelle autonomiche, una distinzione offuscata dalle recenti scoperte che rivelano che gli assoni somatici/nocicettivi hanno effetti efferenti e trofici precedentemente considerati autonomici,8 e che gli assoni, innervando le ghiandole sudoripare e i vasi sanguigni, esprimono il recettore del dolore TRPV1/capsaicina.9
La diagnosi di SFPN è difficile perché i segni familiari di neuropatia a grandi fibre sono assenti o minimi e il test elettrodiagnostico è insensibile. I test raccomandati per la diagnosi obiettiva di SFPN sono stati applicati qui: biopsia cutanea specificamente della gamba distale, immunomarcata per rivelare le fibre nervose epidermiche nocicettive (Epidermal Nerve-Fibers, ENF) 10 (raccomandazione di livello C dell’American Academy of Neurology, raccomandazione di livello A della Federazione Europea delle Società di Neurologia), 11,12 e test di funzionalità autonomica (Autonomic Function Testing, AFT).
Questo consiste in 4 test validati della funzione cardiovagale, adrenergica e sudomotoria delle piccole fibre (raccomandazione di livello B dell’American Academy of Neurology) .12,13
A, Paziente con sudorazione ritardata del tronco e delle braccia durante il test del sudore termoregolatore. Questo ragazzo di 20 anni ha avuto eritromelalgia non dipendente dalla lunghezza (dolore peggiorato dal calore, più arrossamento e gonfiore) che era peggiorato nelle guance e nelle orecchie, ma anche nelle mani e nei piedi, frequenza cardiaca e pressione sanguigna labili e diarrea. La sua biopsia cutanea della gamba distale presentava anomalie minori e anticorpi antinucleo erano presenti in un titolo di 1:80.
Il test della sudorazione termoregolatoria comporta l’applicazione di un indicatore rosso di Alizarina sulla pelle prima del riscaldamento controllato; è arancione quando è asciutto e diventa viola quando è bagnato. Quando eseguito presso la Mayo Clinic, questo ha rivelato una sudorazione ritardata del tronco e delle braccia rispetto alle mani e alle cosce. I risultati degli AFT di questo paziente erano normali alla Mayo Clinic, ma anormali alla Cleveland Clinic e al National Institutes of Health, con una ridotta sudorazione a livello degli avambracci e dei siti di studio distali delle gambe.
I suoi sintomi a chiazze/prossimali e i risultati del test erano coerenti con la neuronopatia/ganglionopatia non lunghezza-dipendente. Immagine gentilmente concessa da Paola Sandroni, MD, PhD, Mayo Clinic.
B, Confronto di genere e sito di produzione di sudore evocato con acetilcolina in soggetti di controllo sani e in pazienti. Tutti i valori dei pazienti (n = 33, 10 ragazzi e 23 ragazze) sono stati confrontati con tutti i valori dei soggetti di controllo abbinati per genere e fascia d’età (n = 38; 19 ragazzi e 19 ragazze). I simboli rappresentano i volumi medi di sudore in ciascun sito di studio ± SEM; le linee collegano gruppi di controlli e pazienti dello stesso sito. La sudorazione era ridotta nei pazienti di sesso maschile rispetto ai soggetti maschili di controllo sull’avambraccio (P = .0011) e sulla gamba prossimale (P = .0017). La sudorazione era ridotta nei pazienti di sesso femminile rispetto ai soggetti femminili di controllo sull’avambraccio (P = .024), sulla gamba prossimale (P = .024) e sul piede (P = .009).
La polineuropatia è stata ritenuta essere rara nei bambini, costituita da casi occasionali di sindrome di Guillain-Barré acuta (Guillain-Barré Syndrome, GBS) e Polineuropatia Demielinizzante Infiammatoria Cronica (Chronic Inflammatory Demyelinating Polineuropathy, CIDP) causata da attacco immunitario su grandi assoni mielinici motori.14 La SFPN, in cui le fibre C di piccolo diametro e le fibre δ-A sono preferenzialmente danneggiate, era poco conosciuta nei bambini ad eccezione di casi genetici rari.14,15 Casi isolati di Eritromelalgia (alias eritermina), un fenotipo storico comprendente dolore bruciante, arrossamento, edema e sollievo con il raffreddamento,16 sono stati descritti nei bambini17 e alcuni casi con esordio prepuberale sono stati collegati a mutazioni del canale del sodio.18,19
La biopsia cutanea è stata la chiave per diagnosticare la SFPN in un bambino giapponese di 12 anni con un nuovo sintomo di dolore diffuso e gastroparesi.20 Noi ed altri abbiamo riferito casi di adolescenti con eritromelalgia acuta e disautonomia in cui la biopsia cutanea confermava una SFPN severa, e il trattamento con corticosteroidi era curativo.21,22 Casi simili reattivi agli steroidi e poi descritti nei bambini più piccoli.23,24 I bambini rappresentano 4 dei 21 casi di una Polineuropatia Acuta delle fibre di piccolo calibro e di fibre di grosso calibro più recente, 25 e recenti studi collegano l’Eritromelalgia Pediatrica nei bambini alla SFPN, proprio come negli adulti.26,27 Diverse nuove relazioni preliminari forniscono prove oggettive che la SFPN è prevalente nella fibromialgia adulta, incluso il nostro studio che identifica la SFPN nel 50% dei pazienti con fibromialgia rispetto allo 0% dei controlli corrispondenti.28-31
Metodi
Selezione di Casi e Dati
Dopo l’approvazione del Comitato di Revisione Istituzionale, sono stati selezionati i documenti ambulatoriali per selezionare i soggetti. I criteri di inclusione erano le cure mediche dell’autore A.L.O. tra aprile 2007 e aprile 2011 per il dolore multifocale diffuso (presente in > 1 arto o regione corporea) che inizia prima dei 21 anni. I criteri di esclusione erano una causa obiettiva identificata del dolore. Abbiamo ottenuto e letto tutti i record disponibili estraendo tutte le relazioni sui fornitori, sui laboratori, sulla fisiologia, sulla patologia e sulla radiologia. Tutti i risultati dei test tecnicamente adeguati dei laboratori accademici o commerciali sono stati inclusi e interpretati come riportati, o utilizzando intervalli di riferimento standard per età.32
Risultato primario: diagnosi obiettiva di SFPN
Non ci sono criteri diagnostici di consenso per SFPN in adulti o bambini,33 quindi abbiamo integrato i risultati di tutti i test diagnostici oggettivi raccomandati; biopsia cutanea immunomarcata con PGP9.5, biopsia cutanea distale, AFT e biopsia sensoriale.11,12 Quesito di SFPN confermato ≥ 1 diagnosi di “SFPN definita” con esame obiettivo. La diagnosi di “SFPN probabile” richiedeva anormalità minori su ≥ 2 test diversi e una “SFPN possibile” richiedeva ≥ 1 anormalità obiettiva minore.
Il test elettrodiagnostico (elettromiografia e test di conduzione nervosa) è stato escluso perché questi test non catturano la funzione delle fibre piccole e il test quantitativo sensoriale è stato escluso perché è un test soggettivo basato sul report soggettivo.34 Una biopsia cutanea definitamente anormale richiedeva la diagnosi di SFPN nel referto o che soddisfacesse i criteri diagnostici standard (densità delle fibre nervose dell’epidermide [ENF] ≤ quinto percentile delle norme di laboratorio).
Anomalie minori della biopsia cutanea comprendevano densità ENF borderline (5,1-15 ° percentile o reperto di eccessivo rigonfiamento/ingrossamento degli assoni).35 Un AFT definitivamente anormale richiedeva la diagnosi SFPN nel referto o rispondente a criteri diagnostici standard (anomalie definite in > 1 dominio). Anomalie minori all’AFT richiedevano reperti di anomalie borderline, lievi, minime o isolate. Una biopsia del nervo definitivamente anormale richiedeva una diagnosi SFPN basata sulla visualizzazione ultrastrutturale della perdita di assoni non mielinizzati.
Il test bioptico cutaneo per SFPN è stato standardizzato.12,36 Un punch da biopsia cutanea da 2 a 3 mm di diametro viene rimosso da un sito anestetizzato sulla gamba distale e quindi sezionato verticalmente e immunomarcato contro antigene PGP9.5, un marker pan-neuronale, per visualizzare le ENF e consentire la misurazione della densità degli assoni (Fig. 1).12,37 Segnaliamo la densità ENF per millimetro quadrato dell’area superficiale cutanea da controllare per lo spessore variabile della sezione cutanea dei diversi laboratori.
La maggior parte delle biopsie cutanee sono state interpretate al Massachusetts General Hospital, le cui norme provengono dalla biopsia di 240 volontari di controllo sani selezionati dai 14 agli 86 anni. Le biopsie dei pazienti < 14 anni (n = 6) sono state normate con quelle di 14 anni di età a causa della mancanza di valori normativi per i bambini più piccoli. L’esecuzione delle biopsie cutanee non ha causato eventi avversi.
Figura 1 – didascalia tradotta in italiano
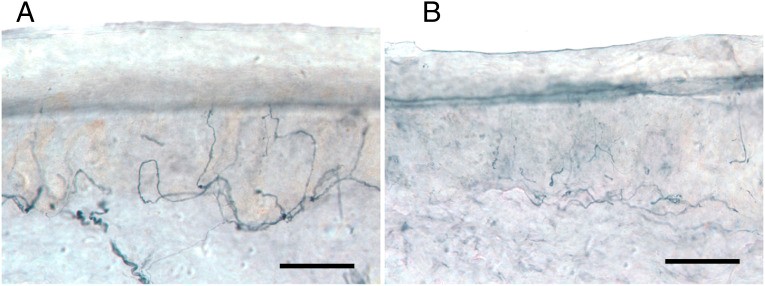
Perdita di fibre nervose immunomarcate PGP 9.5 nelle sezioni verticali dalla biopsia cutanea della gamba distale.
A, La biopsia di un maschio bianco di 19 anni sano di controllo contiene abbondante innervazione (675 ENF/mm2 area di superficie cutanea). B, La biopsia di un paziente bianco di 19 anni dimostra riduzione delle fibre nervose epidermiche (155 ENF/mm2) e dermiche. La SFPN è stata confermata dalla biopsia cutanea; la densità delle ENF = 1.3° percentile dei valori di riferimento normativi di laboratorio. La barra nera rappresenta 50 μm.
L’AFT, considerato più sensibile della biopsia cutanea,39 ha già rilevato SFPN autonomica in metà degli adulti con POTS.40 Poiché i valori normativi per l’AFT provengono da adulti, 41 abbiamo ottenuto il permesso di eseguire un revisione istituzionale per reclutare individui sani di età ≥ 6 anni abbinati per età e genere, per fornire controlli per lo studio pediatrico. Gli intervistati con condizioni potenzialmente neuropatiche sono stati esclusi, e quelli > 18 anni sono stati sottoposti a test di tolleranza al glucosio orale di 2 ore e sono stati esclusi per qualsiasi anormalità.
Abbiamo utilizzato metodi diagnostici, attrezzature (WR Medical Electronics, Stillwater, MN) e interpretazioni standard.12,41 Abbiamo misurato la variabilità della frequenza cardiaca durante la respirazione profonda (6 respiri al minuto in posizione supina) e la manovra di Valsalva, risposte emodinamiche durante la posizione ortostatica inclinata a 80 ° con la testa in alto e la sudorazione evocata con acetilcolina. Gli intervalli di riferimento (RR) forniti dal produttore definivano la normalità delle risposte alla manovra di Valsalva.
Per la variabilità della frequenza cardiaca, i valori <2.5° centile sono definiti anormali. Per i test di inclinazione, era anomala una caduta sistolica della pressione arteriosa > 20 mm Hg, un calo della pressione arteriosa diastolica > 10 mm Hg, 42 e/o un aumento della frequenza cardiaca ≥ 40 (≤ 18 anni) o un aumento ≥ 30 battiti al minuto (> 18 anni) entro 3 minuti dall’inclinazione.43 Per il test del sudore, l’anormalità è stata definita come un valore al di fuori dell’intervallo di confidenza del 95% delle norme specifiche per sito, sesso ed età.41 L’esecuzione dell’AFT non ha causato eventi avversi.
Analisi statistica
Le statistiche descrittive, presentate come medie ± SD, sono state utilizzate per confrontare gli attributi tra i gruppi. La statistica χ2 ha valutato le differenze tra i gruppi in proporzioni di risultati anormali. La produzione di sudore su quattro siti è stata confrontata tra pazienti e controlli utilizzando test t a 2 code con correzione a più campioni per l’errore di tipo I assumendo α = 0,05 e r = 0,5; quindi, P <.025 ha definito il significato.
Risultati
Quarantuno pazienti consecutivi erano idonei e sono stati inclusi. L’età media all’esordio dei sintomi era di 12,3 ± 5,7 anni; l’età alla presentazione era in media di 20,8 ± 9,1 anni. Le caratteristiche demografiche erano notevoli per la predominanza femminile (73%; P = 0.001) e la diversità etnica e geografica. Tre bambini vivevano in Ecuador, Svizzera e Trinidad, e uno aveva sviluppato sintomi prima dell’adozione dall’Estonia all’età di 6 anni. Tra i pazienti di origine americana, 1 aveva 2 genitori coreani e 1 era per metà libanese e per metà bianco americano.
Esito dei test diagnostici per SFPN
Complessivamente, il 59% (24/41) dei pazienti ha soddisfatto i criteri per la SFPN definita, sebbene nessuno di essi fosse stato sottoposto a tutti i 3 test analizzati. Nello specifico, il 30% (11/37) delle biopsie cutanee, il 100% (2/2) delle biopsie del nervo e il 53% (18/34) dei AFT sono stati diagnosticati con “SFPN definita”. Anomalie minori riportate nell’81% delle rimanenti biopsie cutanee (22/26) e 93% del rimanente AFT (14/15) hanno identificato un ulteriore 17% di pazienti con “SFPN probabile” e il 22% con “SFPN possibile”.
Solo 1 su 41 pazienti ha avuto risultati del tutto normali. Due su due, biopsie muscolari e del nervo, hanno diagnosticato SFPN basata su una grande quantità di cellule di Schwann che erano assoni rigeneranti isolati vuoti o contenuti. I nervi sottoposti a biopsia mancavano di demielinizzazione, perdita di grandi fibre, infiltrati infiammatori, vasculite o amiloide. Un muscolo era normale e l’altro aveva cambiamenti neuropatici e da disuso (inattività o scarsa attività muscolare N.d.T)
Il confronto dei risultati AFT tra pazienti e controlli normali ha rivelato nel 27% dei pazienti (vs 3% dei controlli, P <0,001) una ridotta variabilità della frequenza cardiaca durante la respirazione profonda, nel 42% dei pazienti risposte cardiovascolari anormali alla Manovra di Valsalva (vs 0% di controlli; P <0,001) e nel 75% risultati anomali al test sul Tavolo di inclinazione (Tilt Test Table, N.d.T.) (vs 18% dei controlli; P <0,001). La produzione di sudore (Fig 2B), considerata la più sensibile tra i test di funzionalità autonomica per la diagnosi di SFPN, 44 è risultata ridotta in uno o più dei 4 siti testati nell’82% dei pazienti (vs 34% dei controlli; P <.001) .
Caratterizzazione di storie mediche, esami e altri test
La maggior parte dei pazienti era malata moderatamente o gravemente, dimostrato dalle loro vaste indagini/valutazioni mediche presso i principali centri medici accademici. Il 68% era stato ricoverato in ospedale e il 68% aveva chiesto di lasciare la scuola o il lavoro. Non sono state identificate altre diagnosi patogenetiche che spiegassero il loro dolore diffuso (ad es., Artrite, miopatia). L’etichetta sindromica più comune all’ingresso nello studio era la fibromialgia; altri includevano disordine funzionale, sensibilizzazione centrale, sindrome da amplificazione del dolore, stanchezza cronica, sindrome del dolore miofasciale e malattia di Lyme cronica sieronegativa.
Le etichette sindromiche specifiche per organo comprendevano POTS, intestino irritabile, dispepsia funzionale, emicrania addominale e cefalea cronica quotidiana. Quattro pazienti avevano anche sindrome dell’ovaio policistico e 3 avevano la sindrome di Ehlers-Danlos. Le diagnosi psichiatriche (somatizzazione, conversione) erano state spesso considerate, sebbene fosse stata documentata solo 1 malattia psichiatrica, in particolare la depressione maggiore attribuita al dolore incessante.
Gli esami generali hanno identificato costantemente solo tachicardia sinusale e pressione arteriosa anomala, di solito ipotensione ortostatica. Diversi pazienti hanno documentato vesciche neurogene (da infiammazione neurogena, N.d.t.) episodiche. Queste erano spesso sugli arti, anche nel cavo orale e sul viso. I pazienti occasionali avevano riflessi pupillari pigri.
La maggior parte dei test, oltre che per la neuropatia, non fornivano contributi utili per la diagnosi. Il monitoraggio cardiaco ha rivelato tachicardia sinusale (5/5) con occasionale bradicardia intermittente (in accordo con la disautonomia neuropatica) e l’ecocardiografia non dava contributi diagnostici. L’elettromiografia (n = 20) non dava contributi, con solo la denervazione dell’arto superiore identificata in 1 paziente studiato durante la plexite brachiale.
Gli studi sulla conduzione nervosa (n = 24) non fornivano contributi diagnostici, con una polineuropatia identificata in soli 2 pazienti: uno con diabete di tipo 1 di lunga data e l’altro con insufficienza renale da sindrome di Goodpasture. Cinque pazienti presentavano anomalie sensoriali borderline o isolate che potrebbero rappresentare un coinvolgimento subclinico delle grandi fibre.
L’endoscopia gastrointestinale era universalmente inutile, ma gli studi di imaging e motilità addominale (cioè, lo svuotamento gastrico e gli studi con marcatori radiopachi di Sitz [Rx dei tempi di transito N.d.T.]) a volte mostravano una motilità rallentata compatibile con la disautonomia neurogenica. L’imaging cerebrale mediante risonanza magnetica (n = 23) e la tomografia assiale computerizzata (n = 8) non sono state diagnostiche tranne in 1 paziente con edema cerebrale ipertensivo fatale ed emorragia associata alla sindrome di Goodpasture. L’imaging spinale (n = 22) non dava contributi diagnostici salvo occasionalmente identificare la stipsi. Gli studi sul sonno e l’elettroencefalografia erano inutili.
Un’ampia valutazione per le cause note di SFPN ha identificato solo 3 diagnosi sistemiche potenzialmente causali o contributive: 2 pazienti avevano disturbi dello spettro di Sjögren e 1 avevano diabete di tipo 1.37 Il diabete si dimostrò non essere la causa della grave SFPN di questo paziente quando ebbe immediato drammatico miglioramento dopo somministrazione di corticosteroidi, seguita da prolungata remissione dopo trattamenti con immunoglobulina endovenosa (IVIG).
Le storie dei pazienti erano complessivamente significative solamente per la prevalenza di malattie autoimmuni nel 33%. Queste erano generalmente organo-specifiche e spesso associate ad autoanticorpi, inclusi 6 pazienti con storie di tiroidite autoimmune e 2 con porpora di Henoch-Schönlein. Uno aveva avuto episodi di plexite brachiale, diabete di tipo 1, artrite post-virale, porpora trombocitopenica immune, morbo di Crohn, trochleite autoimmune e encefalopatia di Hashimoto.
Questi, insieme con le storie familiari di autoimmunità nel 52%, hanno suggerito la possibilità di cause disimmune di SFPN ad esordio giovanile. Non c’erano storie familiari di SFPN, anche se 1 paziente aveva più membri della famiglia etichettati con fibromialgia che successivamente hanno ricevuto diagnosi di SFPN dal nostro laboratorio, coerenti con SFPN familiare non riconosciuta.31
Il 61% percento di pazienti / famiglie ha attribuito l’esordio della propria malattia a precedenti infezioni o lesioni. Il 25% ha documentato precedenti infezioni tra cui Mycoplasma pneumoniae, Bordatella pertussis, Mycobacterium tuberculosis, Streptococco di gruppo A, mononucleosi, influenza e parvovirus. Tra le 11 lesioni documentate che hanno preceduto l’insorgenza del dolore diffuso, 10 sono state lesioni agli arti (ad es. fratture, distorsioni, congelamento) caratterizzate da dolore eccessivo e gonfiore interpretato come Sindrome da Dolore Regionale Complesso (Complex Regional Pain Syndrome, CRPS). Questi avevano ampiamente risolto ed erano distinti dalle malattie del dolore più diffuso e multifocale studiate qui.
Tra i test sui fluidi corporei estesi dei pazienti (Tabelle 1 e 2), 2), le uniche anomalie consistenti erano immuno-correlate: elevata Velocità di EritroSedimentazione (VES), anticorpi antinucleo (ANA) e bassi livelli di componenti del Complemento C3 e C4. Complessivamente, l’89% dei pazienti ha avuto ≥ 1 di queste anormalità. Una prevalenza del 26% di proteina C-reattiva (PCR) alta è stata considerata troppo aspecifica per ulteriori analisi. Crioglobulinemia di tipo III (IgG, IgM e C3) senza epatite è stata identificata in 1 paziente (5%) insieme a ipocomplementemia profonda e alti valori di VES (Fig. 3).
Queste anomalie si sono risolte dopo il trattamento con prednisone. Pazienti sparsi erano stati testati per autoanticorpi associati a polineuropatie somatiche/dolorose delle piccole fibre (periferina, complesso del canale del potassio voltaggio-dipendente e canali del calcio di tipo N) 45-47: su 15 testati, 1 presentava autoanticorpi anti-periferina, 1 su 7 aveva autoanticorpi complessi anti-canale del potassio voltaggio-dipendenti e 1 su 11 autoanticorpi anti-canale del calcio di tipo N.
Assenti autoanticorpi associati a polineuropatie delle grandi fibre e polineuropatie autonomiche pure (ad esempio autoanticorpi del recettore gangliare dell’acetilcolina). Le biopsie cutanee di due pazienti sono state sottoposte ad analisi dermatopatologica esterna inclusa l’immunofluorescenza diretta (Fig. 3). Entrambi contenevano depositi significativi di C4d, C5b-9 e IgM senza infiltrati infiammatori; uno aveva anche un deposito di IgA.
TABELLA 1
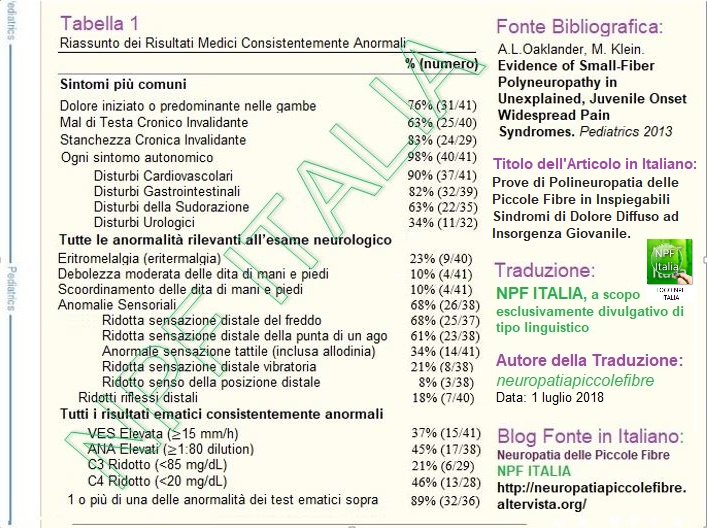
TABELLA 2

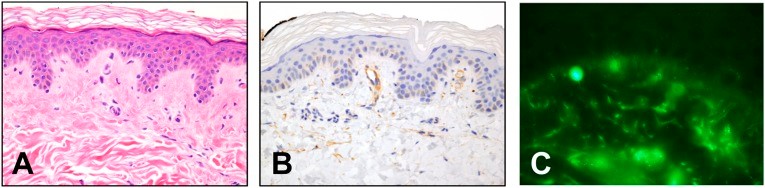
Questa ragazza di 19 anni presentava dolore diffuso, tachicardia, ipotensione ortostatica, mal di testa, nausea e stipsi fin dall’infanzia. Gli esami del sangue hanno identificato crioglobulinemia di tipo III (IgG, IgM, C3), nessuna epatite, basso complemento (C4 = 6, 7, 11, RR, 16-38 mg / dL, C3 = 35, 52; RR, 86-184 mg / dL; VES = 20-26; RR, 0-17 mm / ora). La SFPN è stata confermata dalla biopsia cutanea; Densità ENF = 2.4° centile (non mostrato). Fotomicrografia per gentile concessione di Cynthia Magro, MD, Weill Cornell Medical College (sezioni verticali, 4 μm, ingrandimento 40 ×).
A, Ematossilina e colorazione di eosina che descrivono gonfiore delle cellule endoteliali e ispessimento della membrana basale che indicano microvasculopatia cronica insidiosa. B, Reattività immunoistochimica dei microvasi dermici per C4d, una componente stabile dell’attivazione classica del complemento e una probabile correlazione del suo basso C4 nel siero. C, Etichettatura immunofluorescenza diretta dei depositi di IgM su microvasi dermici, un marcatore aspecifico di microvasculopatia coerente con la deposizione degli immunocomplessi IgM presenti nel suo sangue
Riepilogo dei trattamenti per i sintomi della disautonomia e del dolore neuropatico
Trattamento guidato dei sintomi dei pazienzi con diagnosi di SFPN. L’ipotensione veniva gestita consigliando maggior assunzione di sale e liquidi, evitando di alzarsi in piedi in modo rapido o restarvi prolungatatamente, contraendo i muscoli delle gambe mentre si è in piedi, sollevando la testa del letto ed evitando di potenziare i farmaci. L’ipotensione refrattaria è stata trattata con calze compressive e busti addominali, midodrina o secondariamente, fludrocortisone.48
La tachicardia cronica è stata generalmente ben tollerata, ma se sintomatica è stata trattata con bloccanti del canale del calcio o beta-bloccanti, facendo attenzione a non peggiorare l’ipotensione. I disturbi gastrointestinali sono stati affrontati con diete ricche di fibre, piccoli pasti, elevando la testa del letto ed evitando il decubito postprandiale. Stipsi e nausea/vomito di solito richiedevano un trattamento con farmaci, e 3 pazienti richiedevano il ricovero in ospedale per la disostruzione fecale.
Per il CWP (Chronic Widespread Pain, Dolore Cronico Diffuso) invalidante, il gabapentin è stato prescritto più spesso e sono stati presi in considerazione triciclici a tipo ammina secondari (nortriptilina e desipramina) solo dopo che la disautonomia era stata esclusa o controllata.49 Gli oppioidi sono stati prescritti raramente per dolore severo incontrollato. Il fenotipo dell’eritromelalgia ha indotto a prendere in considerazione la mexiletina.
Riepilogo dei trattamenti con terapie immunomodulanti
L’immunomodulazione è stata considerata per i pazienti con sintomi invalidanti che erano refrattari alla gestione conservativa. Complessivamente, è migliorato l’80% (12/15) tra i pazienti trattati con corticosteroidi e/o immunoglobuline. In assenza delle Linee guida, le dosi sono state estrapolate dai pazienti giovani con GBS (Guillain-Barré Syndrome) o CIDP (Chronic Inflammatory Demyelinating Polineuropathy, Polineuropatia Demielinizzante Infiammatoria Cronica) e casi clinici di SFPN autoimmune.21,22,50,51
Corticosteroidi giornalieri a breve termine sono stati considerati in primo luogo sulla base di basso costo, ampia disponibilità, somministrazione orale e basso rischio di diverse complicazioni nei pazienti giovani. I corticosteroidi (efficaci per CIDP, ma non per GBS52) sono stati associati a un miglioramento prolungato documentato nel 67% (10/15) dei pazienti e sono stati associati a 1 evento avverso maggiore; il paziente diabetico ha sviluppato cataratta. La successiva riduzione del suo prednisone ha causato una ricaduta di SFPN, quindi è stata sottoposta all’IVIG con un recupero graduale sostenuto come documentato di seguito.
Tra 5 pazienti ospedalizzati gravemente malati, trattati con metilprednisolone per via endovenosa (1 g / die per 5 giorni) seguito da uno scalaggio con prednisone, i 2 casi acuti (<3 mesi) incluso il caso indice22 hanno avuto un miglioramento rapido, continuo e oggettivamente documentato (vedi sotto), mentre i 3 pazienti che erano stati ammalati per diversi anni non hanno risposto. Tra i pazienti clinici trattati con prednisone orale (1 mg / kg / die per 4 settimane seguiti da rapido scalaggio), 8 erano migliorati e 2 no.
Il trattamento con IVIG 53 (Intraenous Immunoglobulin) è stato provato in 11 pazienti che non rispondevano ai corticosteroidi o erano responsivi agli steroidi, ma richiedevano un trattamento di lunga durata. Tre non hanno ricevuto una dose di trattamento sufficiente per l’analisi (≤ 3 dosi di 2 g / kg). Tra gli 8 trattati con il regime standard per la polineuropatia autoimmune (≥ 3 volte con 2 g / kg / mese), 3 (38%) non hanno risposto e hanno interrotto il trattamento e 5 (62%) hanno documentato un miglioramento significativo e un trattamento continuato. I tipici sintomi correlati all’infusione hanno risposto ai trattamenti standard. È stato risolto un evento avverso significativo (rash + trombosi venosa profonda). Un paziente ha avuto una ricaduta dopo lo scalaggio IVIG e ha richiesto alcuni trattamenti aggiuntivi.
Il test obiettivo è stato ripetuto per monitorare l’efficacia del trattamento. Sei su 6 Test della Funzionalità Autonomica (Autonomic Test Function, AFT) ripetuti tra i pazienti trattati con terapie immunomodulanti hanno documentato un miglioramento, mentre 2 AFT su 2, ripetuti nei pazienti non immunomodulati, non sono migliorati. Due pazienti immunomodulati, studiati 3 volte ciascuno, hanno avuto un miglioramento al Tilt Test e alle risposte della sudorazione e della variabilità della Frequenza Cardiaca durante la respirazione (13,6 → 14,0 → 19,1 e 4,0 → 9,2 → 14,7 battiti per minuto, rispettivamente). Il 1° paziente immunomodulato, con un basso rapporto Valsalva all’esordio, si era normalizzato (1,42 → 1,76 → 2,27).
Quattro delle 4 ripetute biopsie cutanee tra pazienti immunomodulati hanno documentato la rigenerazione assonale, che è rimasta indietro rispetto al miglioramento dei sintomi. Il paziente indice con area cutanea 0 ENF / mm2 prima del trattamento con corticosteroidi ha avuto 278 ENF dopo 4 anni.22 Un bambino di 10 anni con 51 ENF / mm2 di superficie cutanea prima del corticosteroide e poi IVIG ha avuto 226 ENF 14 mesi dopo. Il paziente diabetico di 19 anni con 0 ENF al basale, due anni dopo ne aveva 48, dopo che l’IVIG aveva prodotto una risoluzione quasi totale. Nel quarto paziente, 4 biopsie nell’arco di 2,5 anni hanno confermato l’inefficacia dei corticosteroidi seguita dall’efficacia di IVIG.
Discussione
Questa serie di casi fornisce nuove ipotesi sulle sindromi di CWP acquisite ad insorgenza nell’infanzia, inspiegabili, implicanti SFPN acquisita, una diagnosi biologicamente plausibile non precedentemente riconosciuta nei bambini. I dati suggeriscono che la SFPN può svilupparsi anche nei bambini in età prescolare e che la SFPN ad esordio giovanile può persistere per decenni nell’età adulta. Tutti i caratteristici pattern temporali (acuto/fulminante versus cronico) e spaziali (distale versus a chiazze/prossimale) delle polineuropatie sono stati tutti identificati.6
Con sintomi disautonomici onnipresenti, segni e anomalie dei test, la SFPN giovanile integra componenti del dolore sia autonomo che neuropatico. Inaspettatamente, il 63% dei pazienti soffriva di mal di testa cronico. Questi non potevano essere attribuiti ad un uso eccessivo di farmaci, in quanto pochi pazienti usavano farmaci per il dolore o il mal di testa. Il mal di testa cronico non è attualmente collegato a ipotensione, microvasculopatia o polineuropatia, 54 ma normalizzare la pressione sanguigna a volte risolveva il mal di testa dei pazienti, coerentemente con la causalità emodinamica.55,56
Lo studio attuale presenta la limitazione intrinseca delle serie di casi da pratiche accademiche, ossia il bias di riferimento, che probabilmente ha aumentato la prevalenza di SFPN. Questa limitazione dovrebbe essere affrontata dalla potenziale rivalutazione di queste scoperte retrospettive in altri contesti.
Rigorosi test su queste famiglie per le cause curabili del dolore cronico dei loro figli hanno generato gli abbondanti dati analizzati qui. I risultati attuali possono aiutare a ridurre i test non necessari in altri bambini simili. L’imaging è stato futile e il test del fluido corporeo ha escluso tutte le cause comuni di SFPN dell’età adulta (Tabella 2). 37,57,58 I test di laboratorio hanno implicato solo marcatori sierologici specifici associati a disimmunità organo-specifica, una causa nota di polineuropatie delle grandi fibre. Questo studio conferma ed estende una serie di 32 casi di eritromelalgia pediatrica che documentano risultati simili, vale a dire SFPN nel 59% dei pazienti e anticorpi antinucleo nel 46% .27 Questo studio non ha misurato il Complemento e non ha riscontrato VES elevata, definiti prudenzialmente come ≥ 30 mm/ora (Mark Davis, comunicazione personale, N.d.T. supervisore dello studio) .27
Nel nostro studio, la sierologia più informativa è stata un basso Complemento C4 nel 46% e livelli basso-normali nella maggior parte degli altri pazienti, in accordo con l’immunità innata e anticorpo-mediata. Questo molto probabilmente coinvolge il percorso del Complemento classico o della Lectina piuttosto che quello alternativo.
Le patologie autoimmuni associate a comorbilità prevalentemente associate agli anticorpi e la mancanza di infiltrati cellulari all’interno delle biopsie del nervo hanno anche argomentato contro il coinvolgimento delle cellule T, sebbene la cellularità all’esordio non possa essere esclusa in quanto le biopsie del nervo sono state eseguite in ritardo nel decorso della malattia.
L’identificazione dermatopatologica del complemento (incluso C4) e depositi di immunoglobuline senza cellularità in 2 biopsie cutanee su 2 (Fig. 3) fornisce un’ulteriore prova di un ruolo per la disimmunità mediata da Autoanticorpi e Complemento nella patogenesi di alcuni casi di SFPN a esordio giovanile.
Il 61% dei pazienti ha segnalato specifici fattori scatenanti di malattia, incluse infezioni, che sono ben noti precipitanti dell’autoimmunità organo-specifica inclusa la neuropatia.14,59 Il 33% ha riferito un antecedente traumatico. Il trauma era stato associato solo raramente alla neuropatia autoimmune60 fino alla recente scoperta fondamentale di neuropatie autoimmuni post-chirurgiche.61 La maggior parte delle lesioni antecedenti tra i nostri pazienti inizialmente causava sindromi dolorose focali etichettate come Sindrome del Dolore Regionale Complesso, anch’essa legata alla Neuropatia delle Piccole Fibre e all’autoimmunità 62-64
Conclusioni
Questo studio ha analizzato i risultati di più test oggettivi per identificare una causa potenzialmente comune per sindromi di Dolore Cronico Diffuso (CWP) ad esordio infantile: la SFPN. Estende il range di età della SFPN alla prima infanzia e caratterizza la presentazione pediatrica. In alcuni pazienti offre evidenza preliminare di immunità disordinata, tra cui l’ipocomplementemia, tra altre anomalie sierologiche, e la sensibilità alla terapia con corticosteroidi e immunoglobuline in alcuni pazienti. Il riconoscimento della SFPN a esordio giovanile dovrebbe aiutare pazienti, famiglie e medici suggerendo un percorso razionale per la valutazione e il trattamento diagnostico.
Avere una diagnosi specifica da testare e curare, quando presente, può ridurre test e trattamenti inefficaci, costosi e potenzialmente dannosi e consentire test oggettivi e il trattamento definitivo di alcuni pazienti. Inoltre, i risultati dimostrano la necessità di valori normativi pediatrici per i test per la SFPN e forniscono nuove ipotesi testabili per lo studio clinico e di ricerca di base.
Ringraziamenti
Ringraziamo i pazienti, le famiglie e i medici per aver fornito i documenti; Heather Downs, BS, e Daniela Herzog, BA, per l’assistenza con i soggetti normali; Vanda Lennon, MD, PhD, per testare sieri per autoanticorpi; e Isabelle Rapin, MD, per una discussione utile. Questo lavoro è dedicato alla memoria di John W. Griffin, MD.
Glossario
AFT Test della Funzionalità Autonomica
CIDP Polineuropatia Infiammatoria Demielinizzante Cronica
CWP Dolore Cronico Diffuso
ENF Fibre Nervose Epidermiche
VES Velocità di Eritro-Sedimentazione
Sindrome di GBS Guillain-Barré (Polineuropatia Demielinizzante Infiammatoria Acuta)
IVIG Immunoglobulina per via endovenosa
POTS Sindrome da Tachicardia Ortostatica Posturale
Intervallo di riferimento RR (dei valori normali per i test di laboratorio)
SFPN Polineuropatia delle Piccole fibre
Note di chiusura
La Dr.ssa Oaklander concettualizzò e progettò lo studio, ottenne finanziamenti, estrasse i dati, partecipò all’analisi dei dati e redasse il manoscritto iniziale e le riscritture. Il dott. Klein ha eseguito il test della funzione autonomica sui soggetti di controllo normali, ha partecipato all’analisi dei dati, ha contribuito alla stesura e alla modifica delle figure, ha contribuito a riscrivere il manoscritto e ha approvato le versioni presentate e tutte le versioni rivedute del manoscritto.
Questo lavoro è stato presentato in forma di Abstract all’American Neurologic Association (25-27 settembre 2011, Manchester Grand Hyatt, San Diego, California) e alla Peripheral Nerve Society (25-29 giugno 2011, Bolger Center, Potomac, MD).
INFORMATIVA FINANZIARIA: Gli autori hanno indicato di non avere relazioni finanziarie rilevanti per questo articolo da divulgare.
FINANZIAMENTO: Supportato in parte dal Servizio Sanitario Pubblico (K24NS059892), dal Dipartimento della Difesa (GW093049) e dai Fondi della famiglia Bradley e Curvey.
Note della Redazione, Ringraziamenti, Citazioni delle Fonti: Questo articolo – in ogni sua parte, inclusa traduzione – è puramente a scopo divulgativo e non è in alcun modo a scopo di lucro, ma pur sempre proprietà intellettuale degli anonimi Autori. Non sostituisce in alcun modo fonti mediche ufficiali, né il lavoro del medico. Rivolgersi sempre al proprio medico e fare sempre riferimento a testi e fonti originali. Non si risponde di alcun utilizzo improprio. Qualsiasi utilizzo a scopo di lucro non è autorizzato.
Per la diffusione di questo materiale si raccomanda la citazione della Fonte mediante pseudonimo dell’Autore in italiano neuropatiapiccolefibre con link alla Home di questo Blog https://neuropatiapiccolefibre.altervista.org/ o link al presente articolo. Si raccomanda inoltre link della Fonte originale in lingua inglese sopra menzionato. Si tratta di un articolo ad accesso libero, che consente l’uso, la distribuzione e la riproduzione illimitati su qualsiasi supporto, a condizione che l’opera sia citata correttamente.
Per citare questo articolo: “IN EVIDENZA” Neuropatia delle Piccole Fibre Bambini e Adolescenti Dolore e Disautonomia | Prove Scientifiche sin dal 2013 https://neuropatiapiccolefibre.altervista.org/neuropatia-delle-piccole-fibre-bambini-e-adolescenti/ Autore in italiano: neuropatiapiccolefibre Blog: https://neuropatiapiccolefibre.altervista.org
Autore dell’Introduzione della Redazione e della Traduzione: neuropatiapiccolefibre
Riferimenti Bibliografici
1. Wolfe F, Smythe HA, Yunus MB, et al. Report of the Multicenter Criteria Committee . The American College of Rheumatology 1990 Criteria for the Classification of Fibromyalgia. Arthritis Rheum. 1990;33(2):160–172 [PubMed]
2. Buskila D. Pediatric fibromyalgia. Rheum Dis Clin North Am. 2009;35(2):253–261 [PubMed]
3. van Geelen SM, Bakker RJ, Kuis W, van de Putte EM. Adolescent chronic fatigue syndrome: a follow-up study. Arch Pediatr Adolesc Med. 2010;164(9):810–814 [PubMed]
4. Mikkelsson M, El-Metwally A, Kautiainen H, Auvinen A, Macfarlane GJ, Salminen JJ. Onset, prognosis and risk factors for widespread pain in schoolchildren: a prospective 4-year follow-up study. Pain. 2008;138(3):681–687 [PubMed]
5. Ojha A, Chelimsky TC, Chelimsky G. Comorbidities in pediatric patients with postural orthostatic tachycardia syndrome. J Pediatr. 2011;158(1):20–23 [PubMed]
6. Gorson KC, Herrmann DN, Thiagarajan R, et al. . Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry. 2008;79(2):163–169 [PubMed]
7. Novak V, Freimer ML, Kissel JT, et al. . Autonomic impairment in painful neuropathy. Neurology. 2001;56(7):861–868 [PubMed]
8. Holzer P. Efferent-like roles of afferent neurons in the gut: Blood flow regulation and tissue protection. Auton Neurosci. 2006;125(1-2):70–75 [PMC free article] [PubMed]
9. Gibbons CH, Wang N, Freeman R. Capsaicin induces degeneration of cutaneous autonomic nerve fibers. Ann Neurol. 2010;68(6):888–898 [PMC free article] [PubMed]
10. McCarthy BG, Hsieh ST, Stocks A, et al. . Cutaneous innervation in sensory neuropathies: evaluation by skin biopsy. Neurology. 1995;45(10):1848–1855 [PubMed]
11. Lauria G, Hsieh ST, Johansson O, et al. European Federation of Neurological Societies. Peripheral Nerve Society . European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol. 2010;17(7):903–912, e44–e49 [PubMed]
12. England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology . Practice Parameter: evaluation of distal symmetric polyneuropathy: role of autonomic testing, nerve biopsy, and skin biopsy (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72(2):177–184 [PubMed]
13. Assessment: Clinical autonomic testing report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1996;46(3):873–880 [PubMed]
14. McDonald CM. Peripheral neuropathies of childhood. Phys Med Rehabil Clin N Am. 2001;12(2):473–490 [PubMed]
15. Axelrod FB, Chelimsky GG, Weese-Mayer DE. Pediatric autonomic disorders. Pediatrics. 2006;118(1):309–321 [PubMed]
16. Mitchell SW. On a rare vaso-motor neurosis of the extremities, and on the maladies with which it may be confounded. Am J Med Sci. 1878;151:17–36
17. Rauck RL, Naveira F, Speight KL, Smith BP. Refractory idiopathic erythromelalgia. Anesth Analg. 1996;82(5):1097–1101 [PubMed]
18. Drenth JP, Finley WH, Breedveld GJ, et al. . The primary erythermalgia-susceptibility gene is located on chromosome 2q31-32. Am J Hum Genet. 2001;68(5):1277–1282 [PMC free article] [PubMed]
19. Dib-Hajj SD, Rush AM, Cummins TR, et al. . Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128(pt 8):1847–1854 [PubMed]
20. Wakamoto H, Hirai A, Manabe K, Hayashi M. Idiopathic small-fiber sensory neuropathy in childhood: A diagnosis based on objective findings on punch skin biopsy specimens. J Pediatr. 1999;135(2 pt 1):257–260 [PubMed]
21. Dabby R, Gilad R, Sadeh M, Lampl Y, Watemberg N. Acute steroid responsive small-fiber sensory neuropathy: a new entity? J Peripher Nerv Syst. 2006;11(1):47–52 [PubMed]
22. Paticoff J, Valovska A, Nedeljkovic SS, Oaklander AL. Defining a treatable cause of erythromelalgia: acute adolescent autoimmune small-fiber axonopathy. Anesth Analg. 2007;104(2):438–441 [PubMed]
23. Pfund Z, Stankovics J, Decsi T, Illes Z. Childhood steroid-responsive acute erythromelalgia with axonal neuropathy of large myelinated fibers: a dysimmune neuropathy? Neuromuscul Disord. 2009;19(1):49–52 [PubMed]
24. Morales PS, Escobar RG, Lizama M, et al. Paediatric hypertension-associated erythromelalgia responds to corticosteroids and is not associated with SCN9A mutations. Rheumatology (Oxf). 2012;51(12):2295–2296 [PubMed]
25. Koike H, Atsuta N, Adachi H, et al. . Clinicopathological features of acute autonomic and sensory neuropathy. Brain. 2010;133(10):2881–2896 [PubMed]
26. Davis MDP, Weenig RH, Genebriera J, Wendelschafer-Crabb G, Kennedy WR, Sandroni P. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55(3):519–522 [PubMed]
27. Cook-Norris RH, Tollefson MM, Cruz-Inigo AE, Sandroni P, Davis MD, Davis DM. Pediatric erythromelalgia: A retrospective review of 32 cases evaluated at Mayo Clinic over a 37-year period. J Am Acad Dermatol. 2012;66(3):416–423 [PubMed]
28. Üçeyler N, Kahn A-K, Zeller D, et al. Functional and morphological impairment of small nerve fibers in fibromyalgia syndrome. In: Proceedings from the 2012 Meeting of the International Association for the Study of Pain; August 27–31, 2012; Milan, Italy. Abstract PT102
29. Solà R, Collado A, Antonelli F, Quiles C, Serra J. Is fibromyalgia a special type of small fiber neuropathy? A microneurography study. In: Proceedings from the 2012 Meeting of the International Association for the Study of Pain; August 27–31, 2012; Milan, Italy. Abstract PW100
30. Shtein R, Hussain M, Hamid M, Raval N, Williams DA, Clauw DJ. In vivo corneal confocal microscopy and clinical correlations in fibromyalgia (FM). In: Proceedings from the 2012 Meeting of the International Association for the Study of Pain; August 27–31, 2012; Milan, Italy. Abstract PH149
31. Oaklander AL, Herzog ZD, Downs HM, Napoleon S. Prevalence of small-fiber polyneuropathy in fibromyalgia. Ann Neurol. 2012
32. Wallach J. Interpretation of Diagnostic Tests. 8th Ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007
33. Devigili G, Tugnoli V, Penza P, et al. . The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain. 2008;131(pt 7):1912–1925 [PMC free article] [PubMed]
34. Freeman R, Chase KP, Risk MR. Quantitative sensory testing cannot differentiate simulated sensory loss from sensory neuropathy. Neurology. 2003;60(3):465–470 [PubMed]
35. Lauria G, Morbin M, Lombardi R, et al. . Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology. 2003;61(5):631–636 [PubMed]
36. Lauria G, Cornblath DR, Johansson O, et al. European Federation of Neurological Societies . EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol. 2005;12(10):747–758 [PubMed]
37. Amato AA, Oaklander AL. Case records of the Massachusetts General Hospital. Case 16-2004. A 76-year-old woman with pain and numbness in the legs and feet. N Engl J Med. 2004;(350):2181–2189 [PubMed]
38. Klein MM, Downs H, Oaklander AL. Normal innervation in distal-leg skin biopsies: evidence of superabundance in youth, subsequent axonal pruning, plus new diagnostic recommendations. Ann Neurol. 2010;68(Suppl S14):S68
39. Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve. 1992;15(6):661–665 [PubMed]
40. Schondorf R, Low PA. Idiopathic postural orthostatic tachycardia syndrome: an attenuated form of acute pandysautonomia? Neurology. 1993;43(1):132–137 [PubMed]
41. Low PA, Sletten DM. Laboratory evaluation of autonomic failure. In: Low PA, Benarroch EE, eds. Clinical Autonomic Disorders. 3rd ed. Baltimore, MD: Lippincott Williams & Wilkins; 2008:130–163
42. Low PA. Prevalence of orthostatic hypotension. Clin Auton Res. 2008;18(suppl 1):8–13 [PubMed]
43. Skinner JE, Driscoll SW, Porter CB, et al. . Orthostatic heart rate and blood pressure in adolescents: reference ranges. J Child Neurol. 2010;25(10):1210–1215 [PubMed]
44. Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve. 2006;34(1):57–61 [PubMed]
45. Chamberlain JL, Pittock SJ, Oprescu AM, et al. . Peripherin-IgG association with neurologic and endocrine autoimmunity. J Autoimmun. 2010;34(4):469–477 [PMC free article] [PubMed]
46. Dhamija R, Renaud DL, Pittock SJ, et al. . Neuronal voltage-gated potassium channel complex autoimmunity in children. Pediatr Neurol. 2011;44(4):275–281 [PubMed]
47. Kimpinski K, Iodice V, Vernino S, Sandroni P, Low PA. Association of N-type calcium channel autoimmunity in patients with autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;150(1–2):136–139 [PMC free article] [PubMed]
48. Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA, Midodrine Study Group . Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. JAMA. 1997;277(13):1046–1051 [PubMed]
49. Dworkin RH, O’Connor AB, Backonja M, et al. . Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237–251 [PubMed]
50. Teasley JE. Initial treatment of childhood chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 2008;38(6):1640–1643 [PubMed]
51. Sladky JT. What is the best initial treatment for childhood chronic inflammatory demyelinating polyneuropathy: corticosteroids or intravenous immunoglobulin? Muscle Nerve. 2008;38(6):1638–1643 [PubMed]
52. Van den Bergh PY, Hadden RD, Bouche P, et al. European Federation of Neurological Societies. Peripheral Nerve Society . European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society – first revision. Eur J Neurol. 2010;17(3):356–363 [PubMed]
53. Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367(21):2015–2025 [PubMed]
54. Classification of the International Headache Society. The international classification of headache disorders, 2nd edition. Blackwell Publishing, Oxford, UK: Cephalalgia. 2004;24(suppl 1):1–160. [PubMed]
55. Mokri B, Low PA. Orthostatic headaches without CSF leak in postural tachycardia syndrome. Neurology. 2003;61(7):980–982 [PubMed]
56. Furlan JC. Headache attributed to autonomic dysreflexia: an underrecognized clinical entity. Neurology. 2011;77(8):792–798 [PubMed]
57. England JD, Gronseth GS, Franklin G, et al. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the AAN, AANEM, and AAPMR. Neurology. 2009;72(2):185–192 [PubMed]
58. Cheng X, Dib-Hajj SD, Tyrrell L, Te Morsche RH, Drenth JP, Waxman SG. Deletion mutation of sodium channel Na(V)1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain. 2011;134(Pt 7):1972–1986 [PubMed]
59. McKhann GM, Cornblath DR, Griffin JW, et al. . Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33(4):333–342 [PubMed]
60. Beghi E, Kurland LT, Mulder DW, Nicolosi A. Brachial plexus neuropathy in the population of Rochester, Minnesota, 1970-1981. Ann Neurol. 1985;18(3):320–323 [PubMed]
61. Staff NP, Engelstad J, Klein CJ, et al. . Post-surgical inflammatory neuropathy. Brain. 2010;133(10):2866–2880 [PubMed]
62. Oaklander AL, Fields HL. Is reflex sympathetic dystrophy/complex regional pain syndrome type I a small-fiber neuropathy? Ann Neurol. 2009;65(6):629–638 [PubMed]
63. Kohr D, Tschernatsch M, Schmitz K, et al. . Autoantibodies in complex regional pain syndrome bind to a differentiation-dependent neuronal surface autoantigen. Pain. 2009;143(3):246–251 [PubMed]
64. Goebel A, Baranowski A, Maurer K, Ghiai A, McCabe C, Ambler G. Intravenous immunoglobulin treatment of the complex regional pain syndrome: a randomized trial. Ann Intern Med. 2010;152(3):152–158 [PubMed]
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti
Neuropatia delle Piccole Fibre Bambini e Adolescenti